病原菌定量基因组
1992 B.A. Hons Mathematical Tripos, University of Cambridge
1998 Ph.D. Department of Biology, University College London
1998-2000 Japan Society for the Promotion of Science Fellow,
Department of Mathematical Biology, Kyushu University
2000-2003 Postdoc, Max Planck Institute for Infection Biology, Berlin
2003-2007 Wellcome Trust Career Development Fellow, Department of Statistics, University of Oxford
2008-2010 Senior Research Fellow, Department of Microbiology,
University College Cork
2010-2014 Senior Scientist, Max Planck Institute for Evolutionary Anthropology, Leipzig
5-7.2014 Project Professor, University of Tokyo
2014-2019 MRC Senior Research Fellow,
University of Swansea and University of Bath
2019-2023 Principle Investigator and Professor,
Institute Pasteur Shanghai, Chinese Academy of Sciences
2023-present Principle Investigator and Professor,Shanghai Institute of Immunity and Infection, CAS
Our coadaptation scans are a form of GWAS, but do not require any phenotype. I see this as a great virtue, because our understanding of what traits are important in bacterial populations is limited by human imagination and the difficulty of recreating realistic conditions within the laboratory. Thus the approach is the ultimate “top down” method to studying variation.
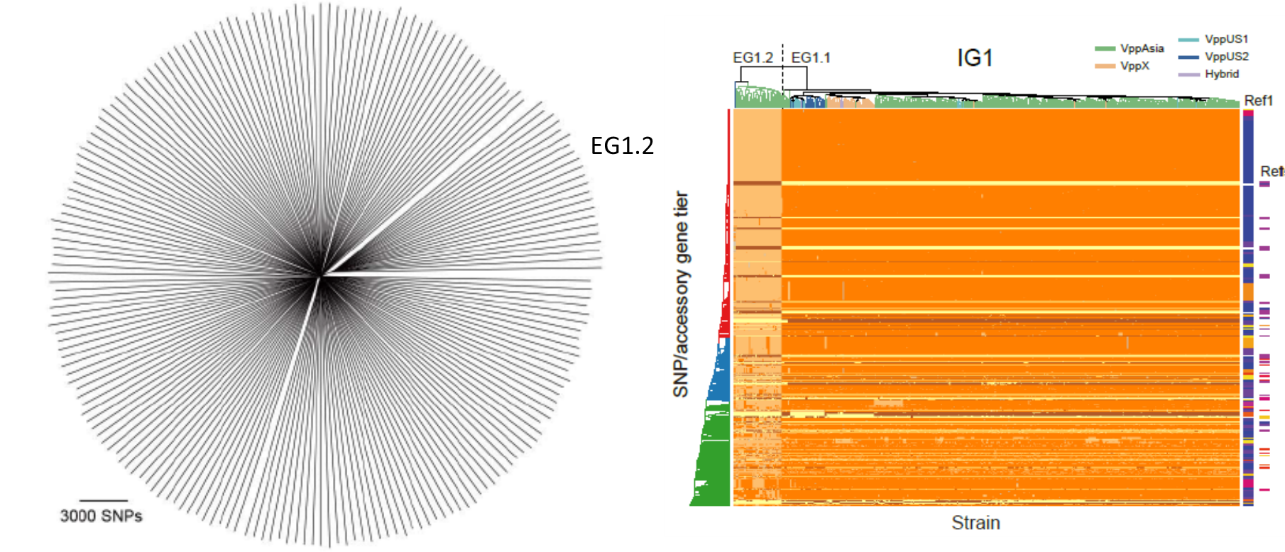
Figure 2. Contrasting patterns of neutral and coadaptive variation in V. parahaemolyticus. Across the genome as a whole EG1.2 strains sharing the same gene pool for >95% of the genome and are barely distinguishable in a neighbour joining tree of genomic variation (A) but more than 100 core and accessory genes, scattered across more than 20 genome regions differ with EG1.1 isolates (B). See our preprint “The landscape of coadaptation in Vibrio parahaemolyticus” https://doi.org/10.1101/373936, for more details.
To date my collaborators and I have used a variety of different approaches to identifying fitness interactions within genomes including Fisher’s exact test, linear mixed models implemented in GEMMA, Direct Coupling Analysis (DCA) implemented in SuperDCA, likelihood-based tests of non-random associations on the phylogeny (unpublished method implemented by Xavier Didelot) and even identified putative interaction signals within components identified in Principal Components Analysis (PCA). Just as important, we have put substantial effort into characterizing the nature of the interactions we have found, displaying them in user-friendly format and interpreting them biologically.
Our experience in these analysis, together with my general experience in analysing genetic data over more than 20 years, suggests that model-based methods can have substantial advantages in terms of statistical power and interpretability but only if the model is appropriate to the set of questions at hand. For example DCA methods have been shown to enhance statistical power in identifying direct interactions between sites that are physically proximal in folded protein molecules. However, in our genome-wide scans, we are interested in identifying multilocus interactions and also identifying hub loci that interact with a large number of others, both of which represent scenarios that deviate substantially from the underlying DCA inference model. Our experience is that interactions that seem of highest interest are sometimes quite low in DCA rankings.
Goals for ongoing methods development include:
· Integrating population based and phylogenetic signals. Using both signals at once will enhance statistical power and robustness as well as making it possible to investigate the dependence of fitness interactions on genomic context.
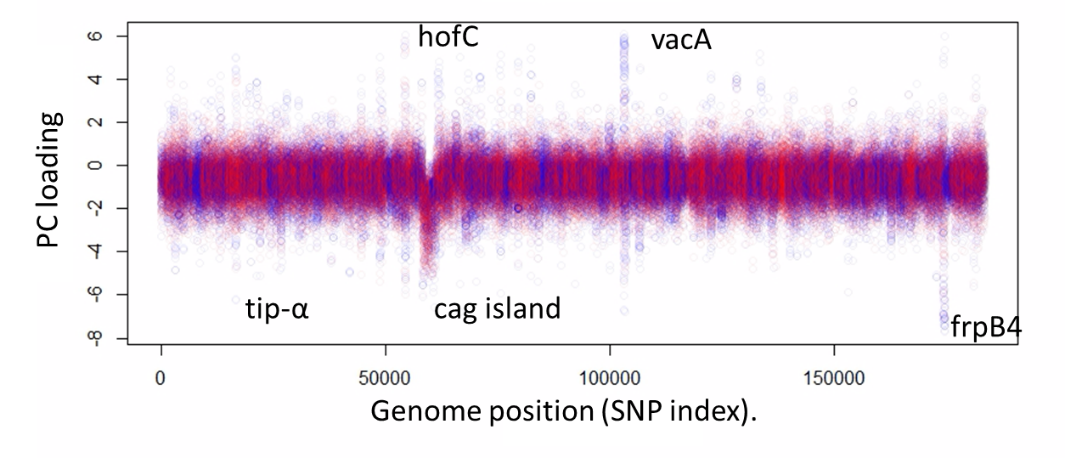
· Identification of quantitative traits without phenotypic measurement. An extremely interesting goal is to detect sets of loci that covary together in response to the underlying trait, without knowing the trait itself as in Figure 3.
· Methods to detect loci that are hubs for many interactions.
· Integrating selection scans with functional and evolutionary annotations.
· Characterization of the pattern of selection maintaining the observed associations.
Figure 3 A virulence associated PC in H. pylori. In H. pylori. This PC is highly enriched for genes involved in virulence as well as iron and/or nickel uptake and appears to represent a quantitative nutrition related trait at the heart of H. pylori virulence biology (unpublished).